


The EU MDR 2020: Deadline for Compliance
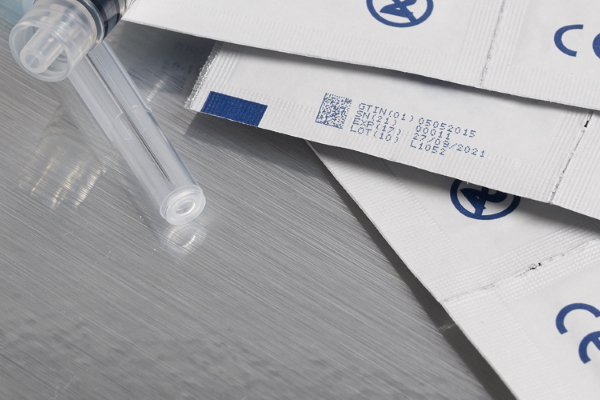
The deadline for ensuring compliance with the European Union’s Medical Device Regulations (EU MDR) is looming. These new, stricter regulations are aimed at improving the traceability features and safety management of medical devices for sale within the EU. With less than nine months to go before the deadline, the countdown is on for medical device manufacturers to ensure they are compliant.
Concerned about what the new regulations mean to you? In this article, Volker Watzke, EU Medical Device Sector Manager at Domino Printing Sciences, offers advice on how to become compliant as quickly and efficiently as possible.
Recap: What is required from the EU MDR?
The EU MDR will come into force on 26th May 2020, replacing the EU’s current Medical Device Directive (93/42/EEC) and Directive on Active Implantable Medical Devices (90/385/EEC).
As part of the new regulations, manufacturers of medical devices for sale within the EU must adhere to strict guidelines to ensure their products are safe to use. The regulations cover all medical devices sold in the EU – everything from scalpels and needles to pacemakers, contact lenses, and prosthetic limbs.
From 26th May 2020, all medical devices will need to be assigned a unique device identification (UDI) code. Devices which fall under Class III and Class IIa/b will need to have their UDI recorded, indexed, and registered on a central EU database called EUDAMED – the European Database for Medical Devices.
Manufacturers of Class III and Class IIa/b products will be responsible for sharing product data according to Annex VI Part B of the regulation. Manufacturers of Class I products will also be required to collect and save product data but need only share this information if requested.
The introduction of the EU MDR obligates medical device manufacturers to invest in technology to enable the fast and accurate application of traceability coding to products and packaging at the individual item level.
Failure to comply with these procedures may mean that devices are withdrawn from sale, with device manufacturers no longer able to supply their products to other EU member states.
Where are we now?
Medical device manufacturers should keep abreast of current developments as and when they arise, and speak to authorities such as GS1, as well as notified bodies and trusted partners for advice and support. Conferences, webinars, and events can offer a wealth of information on how best to manage your procedures and prepare for the new regulations.
At the time of writing, the technical specification for EUDAMED is expected to be released by the end of 2019. So far, manufacturers have the benefit of a technical bulletin, available from the European Commission, which provides information on how data should be submitted. The technical bulletin is addressed to the different needs of each manufacturer.
Manufacturers will have approximately 5-6 months from the release of the technical specification of EUDAMED and the final date of registration. It is, therefore, advisable to begin collecting data as soon as possible. Manufacturers will need to collect data on each product according to the Annex VI, Part B of the EU MDR, and begin preparing the data for sharing on EUDAMED.
As well as preparing all data in advance of the May 2020 deadline, manufacturers should ensure that they have the right partners who support them through the process. To ensure that their devices comply with the new regulations, manufacturers should speak to their code issuing agency and notified bodies for advice.
At present, some large medical device manufacturers are utilising up to 25 per cent of their employee base in bringing their procedures up to standard. Small and medium-sized manufacturers are unlikely to have the capacity to dedicate so much of their workforce and should consider options for external support.
Issuing agencies
In June 2019 GS1 became the first issuing agency for EU MDR compliant codes, meaning that 2D Data Matrix and GS1-128 codes can be used going forward. It is expected that other issuing agencies will follow suit, with potential for the use of HIBCC and ICCBBA coding in the future.
Notified bodies
As part of existing EU Directives (90/385/EWG) and (93/42/EWG) manufacturers audit and check their products on a regular basis to ensure compliance. Notified bodies support manufacturers in this process to ensure that new and existing products can be sold. Of the 57 notified bodies across Europe dealing with current legislation, only 38 have applied for accreditation to the EU MDR.
At the time of writing, only four notified bodies had achieved the new accreditation.
Manufacturers can decide which notified body they choose for their products but should bear in mind that some existing notified bodies will no longer be available for assistance under the EU MDR. The new process of accreditation is significantly more challenging, and as such, around 20 notified bodies have not applied.
The sudden lack of notified bodies has become a bottleneck for manufacturers seeking accreditation of their products and can be seen as a roadblock for innovation, as start-ups struggle to find support. Start-ups looking to register their products should speak to one of the four newly accredited notified bodies for advice.
A global effect
The EU MDR covers all items sold within the EU, but this does not mean that only EU member states need to fulfil the requirements. All manufacturers that wish to sell their product in the EU need to ensure that they satisfy the EU MDR requirements, or they may see their products removed from sale.
Equally, manufacturers who are registered according to the EU MDR should focus future attentions on ensuring compliance in different parts of the world where additional legislation may be a factor. At present, the US, India, and South Korea are in the process of passing legislation on the identification of medical devices and drugs, with further discussions currently in progress in Australia, Brazil, Canada, China, Colombia, Japan, New Zealand, Russia, Saudi Arabia, Singapore, and Turkey.
Who can offer further advice and support?
With the deadline for ensuring compliance with the EU MDR fast approaching, the time to act is now.
The experts at Domino are on hand to help you ensure that your products are compliant with the upcoming EU MDR. At Domino, we provide validation packages to help manufacturers authenticate their products and prepare for EU MDR accreditation.
Our range of coding and marking solutions offers manufacturers of medical devices the most suitable technology in order to achieve UDI compliance. For advice and support in how to ensure your coding is EU MDR compliant, contact the experts at Domino.
For further insight into the upcoming EU MDR, listen to Domino’s latest podcast https://go.domino-printing.com/medicaldevicespodcast

Volker Watzke is EU Medical Device Sector Manager at Domino Printing Sciences
Related articles:
Want our news sent directly to your inbox?

© SecuringIndustry.com
Sponsored Content
Could you take SecuringIndustry.com forward to new heights?
After more than 15 years of publishing SecuringIndustry.com, its co-founders have decided to move on to other projects, and it is time to pass the brand and publication on to new owners who can take it forward editorially and commercially. If you would like more information about the opportunity, please contact us at info@securingindustry.com.
Sponsored Content
Could you take SecuringIndustry.com forward to new heights?
After more than 15 years of publishing SecuringIndustry.com, its co-founders have decided to move on to other projects, and it is time to pass the brand and publication on to new owners who can take it forward editorially and commercially. If you would like more information about the opportunity, please contact us at info@securingindustry.com.
Press Releases
-
》 LSPedia Awarded National DSCSA Compliance Agreement with Premier, Inc
-
》 Biocair looks to the future of drug development and distribution with a new process for risk mitigation in supply chains
-
》 Systech launches innovative exceptions handling and rework solution
-
》 Metrc announces new track-and-trace government contract with the Commonwealth of Kentucky
Partners